We will illustrate Hyperchem Hartree-Fock calculations with a simple example using the sodium (Na) atom. We first create the system ("Build" "Default Element") and then select "Setup" and "Ab Initio" to specify the type of calculation to be performed. In the following example, I am using a "Large" basis set, since this is a simple 1-atom system for which computing time is not an issue. We will need to select "UHF" (unrestricted Hartree-Fock) for the neutral Na atom, since it has an unpaired electron. The spin multiplicity for a single unpaired electron is 2x(1/2)+1 = 2. In the "Advanced Options" dialog, the "Direct SCF calculation" button controls whether electron integrals are recalculated "on the fly" or stored on disk; there may be disk permissions issues if you leave it unchecked.
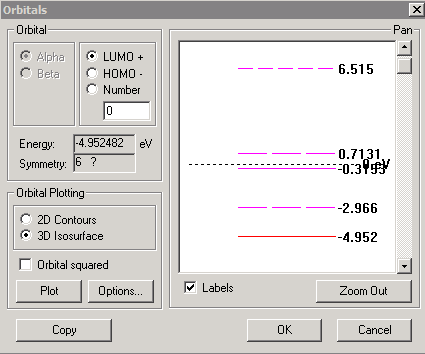
We are now ready to perform the calculations, by selecting "Compute" "Single Point" - after a short time the calculations have converged, and the energy is reported as -101557 kcal/mol. We can select "Compute" "Orbitals" to see the details. In the screenshot below, I have selected the "labels" option to show energy levels and electron occupancies, but this makes the energy level diagram rather cluttered near the top, as the energy of the lowest (1s) level is much lower than for the rest of the orbitals. You can correct this by zooming into a region of the diagram. To do this, select the region of interest with the mouse (see second screen shot below); you may need to do this a few times to get an uncluttered diagram. The Highest Occupied Molecular Orbital (HOMO) state is at -4.961 eV and contains the unpaired electron, as shown below. The Lowest Unoccupied Molecular Orbital (LUMO) state for the alpha electron is at 0.7748 eV.

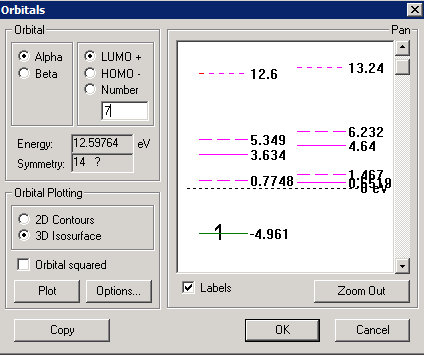
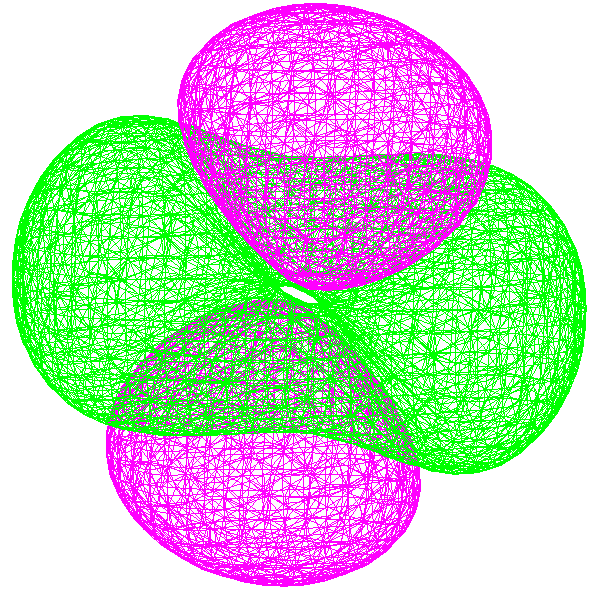
You can select a specific orbital to plot by clicking on it, or by specifying its order in the box below "Number" - note that you specify orbitals relative to the HOMO or LUMO state. In the example above, I have selected the first 3d orbital (LUMO + 7 level) at an energy of 12.6 eV. There are several options that control rendering of the orbitals, whether the molecule surface is shown or not, and the grid size and its spatial extent. In the screen shots below, I have selected a "Wire Mesh" rendering at the 0.01 level (a higher level makes the orbitals appear smaller); I have opted to "Hide molecule" since the atom surface obscured the orbital structure, and I have selected a user-defined grid of sufficient size to prevent clipping of the orbitals at the edge. The orbitals can be rotated in 3D using the standard Hyperchem controls to get a better feeling for their shape.

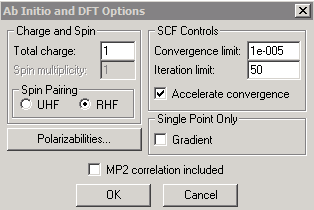
The energy levels of the sodium cation (Na+) can be calculated in a similar way. The only change needed to do this is to specify a charge of +1 in the "Options" panel of the ab initio calculations, as shown below. Since the Na+ cation does not have an unpaired electron, the faster restricted Hartree-Fock (RHF) option can now be selected.
Note that the resulting levels are quite different for the 3p and higher orbitals (see diagram below). The energy difference between 3s and 3p from the Hyperchem calculations is 4.952-2.966 = 1.986 eV (or 624 nm). Experimentally, the 3s <-> 3p transition is a close doublet near 589 nm, resulting in the bright yellow light of sodium lamps.