Hyperchem can perform molecular dynamics simulations under periodic boundary conditions. The easiest type of simulation to set up is one in which the periodic box contains water molecules, in addition to one or more solute molecules of interest. To start an example calculation, let's create a single water molecule in our system:
We can now setup a periodic box around this "solute" molecule:
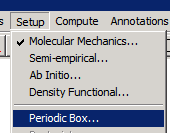
The dialog that appears requests the x,y, and z dimensions of the periodic box, and the "minumum distance" between water molecules and solute.
Once "ok" has been clicked, the periodic box with added water molecules appears:
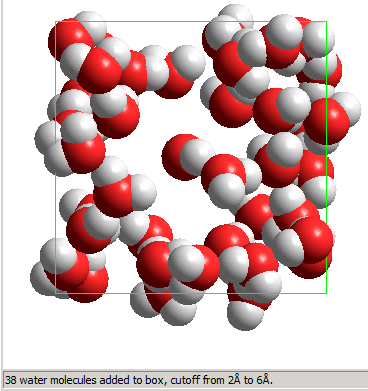
In this particular case, 38 water molecules were added; the number is determined by packing constraints (avoiding overlaps leading to very high energies).
We are almost ready to start the molecular dynamics simulation. The two control screens are shown below, and are used to specify simulation parameters and plotting (or recording) of quantities during the simulation. A time step of 0.5 fs is usually small enough to ensure simulation stability for most systems. A larger simulation time step is advantageous for reaching longer times in less CPU time.
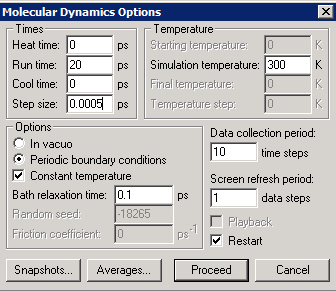

If we attempt to run at this point, an error message appears:
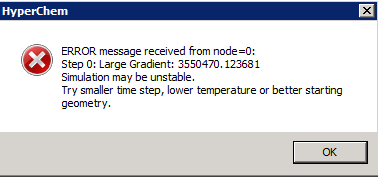
The problem is that the initial configuration still contains unfavorable contacts; it needs to be "annealed" by a cycle of energy minimization before starting the simulation. Once this has been done, the simulation can proceed. Note the quick relaxation away from the initial state, and the slow relaxation at longer times - for this system, equilibrium has not yet been reached after 20 ps - can you guess why it takes so long? [Hint - compute the density]. Also note that the kinetic temperature (green trace) still fluctuates, even though this is a "constant-temperature" simulation.

To perform simulations of systems other than water, We need to set up a periodic box containing just the molecules of interest, which can be done as follows. We start by creating a couple of, e.g., Ar atoms in an empty workspace. Here, I have used the MM+ force field, and "optimized" the geometry of the dimer, to illustrate the potential acting between them (shown in the inset). I have also adjusted the apparent size of the spheres representing Ar, to better illustrate the size of Ar atoms.

Now we again set up a periodic box (this time 18x18x18, which Hyperchem fills with water), but now we select and delete all the water molecules (this can be done quickly by selecting first the Ar atoms, and then using "Select" "Complement Selection"). This leaves us with two Ar atoms in a periodic box. We then insert two Xe atoms (yellow), and then "copy and paste" until we have reached a desired number of atoms in the box. Don't worry if the atoms appear to be outside the box; a cycle of geometry optimization will bring them in the periodic box, and a molecular simulation can be initiated. The snapshot below is from a simulation of a 1:1 mixture of Ar+Kr, using the following simulation parameters:
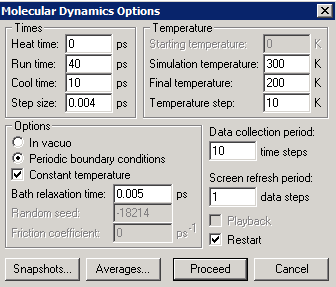
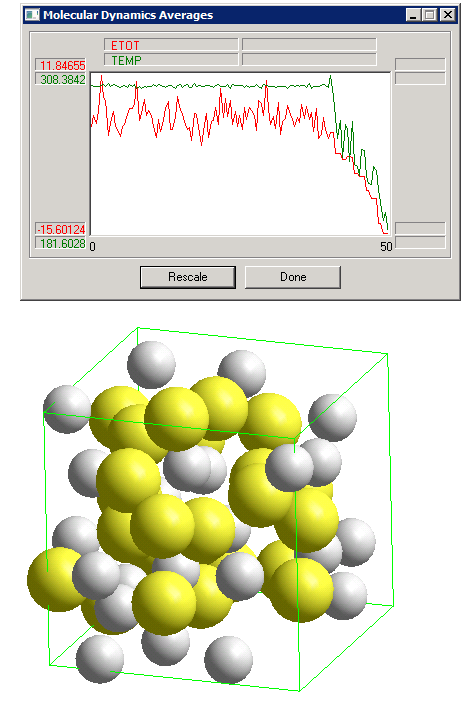
The "Bath relaxation time" parameter of 5 fs strongly suppresses fluctuations in the kinetic temperature. If, on the other hand we used a value of 50 fs, the kinetic temperature fluctuates in a way that more closely approximates the correct behavior for the statistical mechanical constant-temperature ensemble.
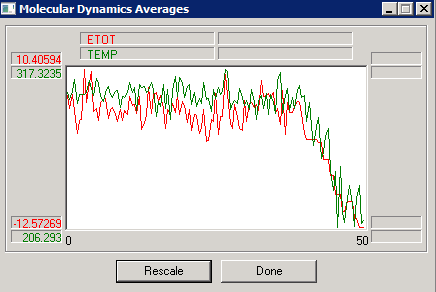
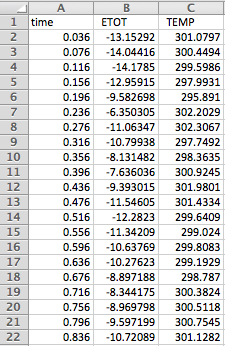
If we wanted to extract data from a simulation in order to analyze them in an external program (e.g., Excel), one of the "Average Only" or "Average and Graph" options should be selected in the "Averages" tab of "Molecular Dynamics Options." At the end of the run, a file with a ".csv" extension (comma-and-space-delimited values) is created in the Hyperchem directory, which can then be imported into Excel for calculations of averages and statistical uncertainties: