
We start our examples with the simplest molecular system, the dihydrogen ion, H2+. After building the system the usual way, we select "Setup" and "Ab Initio" and specify a "Minimal" STO-3G basis set, since this is a simple one-electron system. We need to specify a charge of +1, doublet state UHF calculations. We can "Compute" "Geometry Optimization" to confirm that the bond length is predicted to be 1.06 Angstrom, in excellent agreement with experiments. Now we can select the whole system and "Compute" "Potential" over the range of 0.4 to 4 Angstrom:
The resulting potential function is shown below.

The potential data values can be extracted and manipulated in an external program (e.g., Excel) as follows. We click on "Properties" on the 1D Potential graph above, and then select the System tab, toggle Save Data, specify a File, "H2+" in this case, and click on Save:

This creates a file in the working directory called "H2+.GSP" - use any text editor to open it and you should see the following:
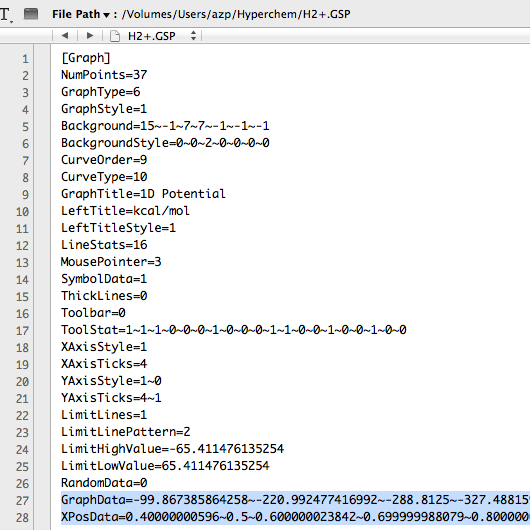
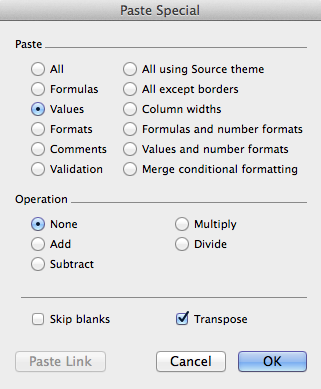
Of particular interest are the last two (long) lines which contain the data; you can copy and paste them into Excel, in order to change units, combine or compare them with other data, etc. For this purpose, it is particularly useful to "transpose" using the following Excel option on "Paste Special:"
I transformed the data into atomic units and plotted them on Excel in order to compare to Fig. 15.3 of Hinchliffe:
