HyperChem is a package for performing Molecular Mechanics, Molecular Dynamics and ab initio calculations for chemical systems.
The first screen that one sees on starting up the package looks as follows:
First, I will describe how to do a simple Molecular Mechanics calculation for a simple molecule, H2O. First, we need to "create" the molecule. This is done by selecting Build : Default Element : and selecting O for Oxygen - the screen looks as follows:

After returning to the main screen, make sure the ![]() button is selected and
click the left mouse button on the main drawing area. You will
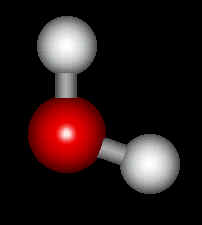
get the following image of an Oxygen atom:
button is selected and
click the left mouse button on the main drawing area. You will
get the following image of an Oxygen atom:

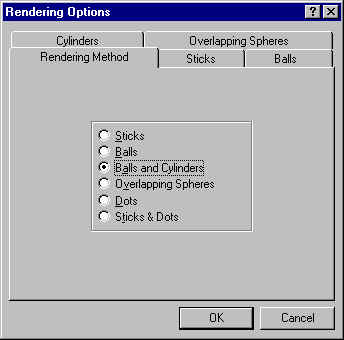
If you do not, you may need to select Display : Rendering Options : Rendering Method : Balls and Cylinders (see screen shot below):
Now select Build : Add Hydrogens and the molecule turns into a water molecule:
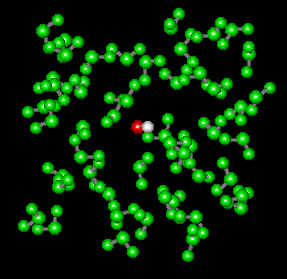
Now "select" the molecule you created by clicking Select : Select All . The molecule should turn green. Now select from the menu Setup : Periodic Box. You will see the following screen:

Enter the desired dimensions in the periodic box (12x12x12 Angstrom in the case above) and click OK. You get a box with 58 "solvent" molecules, which happen to be water in this case. Select from the menu Select : Complement Selection and the image inside your drawing area should look as follows:
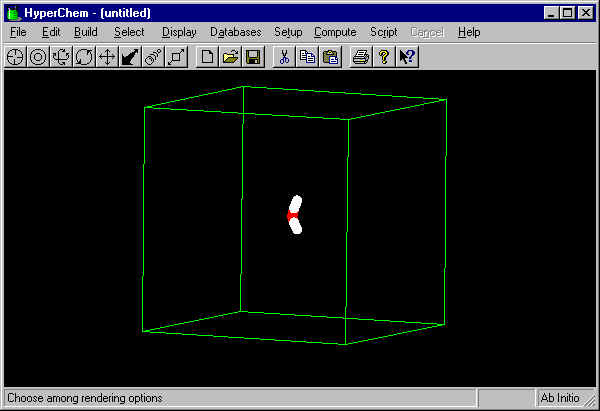
Now delete all the added solvent molecules by pressing the
"Delete" on the keyboard. You can verify that there is
a box now by setting the Rendering mode (see above)
to "Sticks" and making sure that the "Show
Periodic Box" Option is checked (under Display). The
screen (after rotating using the ![]() tool) looks like this:
tool) looks like this:
Now select the water atom and duplicate as many times as you want using Edit : Copy : Past (or CTRL-C CTRL-V).
To setup a Molecular Dynamics Calculation using the MM+ force field, select Setup : Molecular Mechanics from the main menu and click on the MM+ force field button:
After this, you are ready to run a simulation using Compute : Molecular Dynamics and filling out the appropriate options on the screen:

HyperChem will likely complain that the simulation is unstable, because of the poor initial configuration (which probably has overlaps and a very high energy). You can fix this by running a cycle of energy minimization by selecting Compute : Geometry Optimization, which will minimize the energy of the selected molecules by shifting them around. You will need to run for some time and collect averages of the energy.
The application of ab initio calculations to simple electronic structure calculations will be illustrated below for the simple case of determining the potential energy curve as a function of bond length for the molecule O2. The first step is to setup two O atoms in the main simulation box, at a distance (in the case below) of 1.18329 Angstrom.

Different distance can be obtained by selecting one of the atoms and displacing it (Edit : Translate). The next step is to "Setup" the ab initio calculations. The following menu pops up:

Select the basis set desired, and also click on "Options" to get to the following menu:

The "Total charge" entry is for the whole molecule (0 in this case) and the "UHF" or "RHF" denote unrestricted and restricted Hartree-Fock calculations, respectively. For molecules such as O2, RHF calculations are appropriate, since there are no lone electrons.
After the calculations have been set up, you can proceed to Compute : Single Point to obtain the total energy of the configuration. To see details on the results of the calculation, you will need to select File : Start Log to keep your calculations in a file of your choice. The following is an section of the log file for the O2 calculations:
ENERGIES AND GRADIENT Total Energy = -93707.5169532 (kcal/mol) Total Energy = -149.332415288 (a.u.) Electronic Kinetic Energy = 94705.2449307 (kcal/mol) Electronic Kinetic Energy = 150.922395831 (a.u.) The Virial (-V/T) = 1.9895 eK, ee and eN Energy = -116385.3430834 (kcal/mol) Nuclear Repulsion Energy = 22677.8261302 (kcal/mol)
The total energy (in kcal/mol) is also shown at the bottom of the screen once the calculations have converged. New calculations are repeated for different distances, and the following table of potential energy versus distance is contructed (for a "small" basis set of (3-21G) - note that there is a way to place selected molecules at specific locations in space.
| Bond Distance, (Angstrom) |
Energy (Mcal/mol) |
| 0.8 | -92.80 |
| 0.85 | -92.97 |
| 0.90 | -93.09 |
| 0.95 | -93.17 |
| 1.00 | -93.22 |
| 1.05 | -93.26 |
| 1.10 | -93.28 |
| 1.15 | -93.29 |
| 1.20 | -93.30 |
| 1.25 | -93.30 |
| 1.30 | -93.05 |
| 1.35 | -93.08 |
| 1.40 | -93.10 |
| 1.50 | -93.14 |
A "Geometry Optimization" gives an equilibrium bond distance of 1.242 Angstrom for the molecule. The behavior of the energy at longer distances is a little strange (can you guess why?). The experimental value for the bond length is 1.208 Angstrom.