Input file
A typical input file looks like this, with the bold characters representing input values. :
Data for second virial coefficient calculation
[1] Enter type of molecules being calculated
'pure fluorine'
[2] Temperature (K):
143.15
[3] Number of group 1 beads.
2
[4] Number of group 2 beads.
2
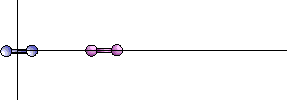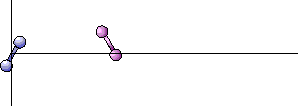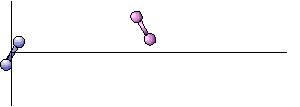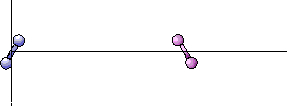
[5] Coordinates and mass of the group1 beads:
TYPE mass x y z
1 14 0.0 0.0 0.0
1 14 1.089 0.0 0.0
Coordinates and mass of the group2 beads:
TYPE mass x y z
1 14 9.0 0.0 0.0
1 14 10.089 0.0 0.0
[6] Number of rotation trials for first to forth interval.
Starting distance (A) Number of rotations.
0 1000
2 1000
6 1000
15 1000
[7] Number of Hamiltonians.
1
[8] Number of LJ groups.
1
[9] Rank 3 array of sigma's(Angstrom) and epsilon/k's(K)
for each hamiltonian. Start new line for new hamiltonian.
sigma 11, 22, ...
3.298
Epsilon 11, 22, ...
36.2
[10] kappa_ij (Correction to Berthelot rule): eps_ij=sqrt(eps_1*eps_j)*(1-kappa_ij)
Enter in order (1,2) (1,3) (1,4) (2,3) (2,4) etc. Start new line for new Ham.
0
[11] lambda_ij (correction to Lorentz rule) : Sig_ij=0.5*(sig_i+sig_j)*(1-lamda_ij)
Enter in order (1,2) (1,3) (1,4) (2,3) (2,4) etc. Start new line for new Ham.
0
[12] Maximum distance between the two groups (Angstrom).
60
[13] Length of normal increments.
0.2
[14] Value of small increments during rapid changes.
0.05
[15] Maximun distance before ending small increments (Angstrom).
15
Details of the Input file.
[1] Type of simulation (can hold up to 20 characters).
[2] Temperature of interest in Kelvins.
[3] Number of atoms in molecule 1.
[4] Number of atoms in molecule 2.
[5] Starting coordinates of molecules 1 and 2. The two could be place anywhere
in 3D space with any separation distance. Setting them on the x-axis is purely for covenience.
[6] Adjust the number of rotations for a given distance. The purpose of this is
that we can increase the number of rotaion where the interactions are significant and decrease
them where changes are not so rapid (such as when the molecules are far from each other). This
greatly reduces the computational time.
[7] Number of Hamiltonians. However, this option is not yet functional. Hamiltonian
equals 1 is used.
[8] Number of Lennard-Jones Groups.
[9] Arrays of epsilon/kb and sigma. Only need values for pure components.
The values for mixtures are later calculated.
[10] Correction to Berthelot rule.
[11] Correction to Lorentz rule.
[12] Maximun separation distance between the two groups (Angstrom).
[13] Longer increments which are used where changes are not so aprupt.
[14] Smaller increments used where changes in the function are rapid.
[15] Distance where transition from small to large increments occur.
Top of Page.
Results File.
A typical output (results) file looks like this:
Results file for second virial coefficient calculation
Calculation for molecule type : pure fluorine
Temperature = 143.1499939 K
Epsilon/k (K) = 36.20000076
Sigma (Angstrom) = 3.298000097
Maximum distance evaluated is at r = 59.85020447 Angstrom
Number of rotations from 0.0000000000E+00 to 2.000000000 = 1000
Number of rotations from 2.000000000 to 6.000000000 = 1000
Number of rotations from 6.000000000 to 15.00000000 = 1000
Number of rotations from 15.00000000 to 59.85020447 = 1000
Number of data points integrated: 525
Small increment = 0.5000000075E-01 Angstrom starts at 0 and changes
to increment = 0.2000000030 at r = 15.05003929 Angstrom
The value of the integral is 19.47787666
The reduced value (B*) of the second virial coefficient is -1.628961682
The value of the second virial coefficient is -73.69910431 cm^3/mol
The reduced value (B*) is defined as :
B* = B(T)/(2/3*pi*sigma3)
Top of page.


{kind=link}