 Gibbs
Project Gibbs
Project
Gibbs
Project Gibbs
Project
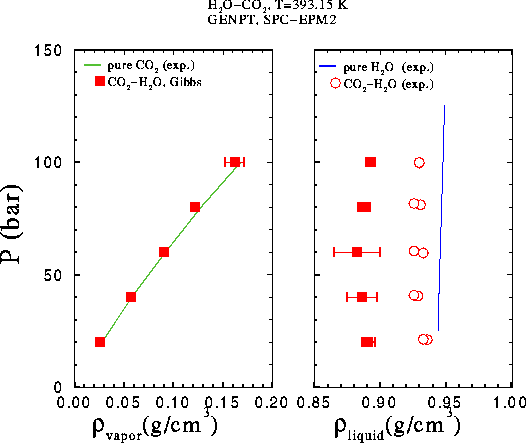
Comparison of simulation data with experimental results. The EPM2 model was used* for carbon dioxide (see also results for CO2), water was modeled using the SPC model (see also results for H2O).
Several points on the phase envelope were calculated using Gibbs1.11 (Theodorou and Suter method for the LJ interactions). Experimental data for the composition of the liquid phase at this temperature were calculated from Wiebe and Gaddy, J. Am. Chem. Soc., 61, 315 (1939), an estimation for the composition of the vapor phase was made using data from Wiebe, Chem. Rev., 29, 475 (1941). Experimental data vor the densities were not available. The system size was N = 250, the runs took from 3 to 4.5 million configurations.
T = 373.15 K, p = 25 bar - 300 bar
Several points on the phase envelope were calculated (Gibbs1.11). Experimental data for the composition of the liquid phase were calculated from values by Wiebe and Gaddy, J. Am. Chem. Soc., 61, 315 (1939) resp. taken from Zawisza and Boguslawa, J. Chem. Eng. Data, 26, 388 (1981), data for the composition of the vapor phase were taken from Coan and King, J. Am. Soc. 93, 1857 (1971) and Tödheide and Franck, Z. Phys. Chem. Neue Folge, 37, 387 (1963). No experimental data for the densities were available.
GEMC-NPT runs with N = 350 particles were carried out and the results compared with data by Nighswander et al., J. Chem. Eng. Data, 34, 355 (1989). The length of the runs was 4 million configurations, all results except the point at 20 bar were obtained using Gibbs0.9 (constant cutoff radius for the LJ interactions), the point at 20 bar was calculated using Gibbs1.11.
CO2-H2O at low temperatures and moderate pressures
The simulation results for the solubility of carbon dioxide in water agree with most of the experimental points for the liquid phase within the statistical uncertainties. Since the influence of the temperature on the gas solubility in water is very small these deviations could not be described by the simulations because the statistical uncertainities cover the whole range of the differences. In the vapor phase the agreement of simulation and experiment in the pressure range up to 100 bar is quite good, the minima of the solubility of water in carbon dioxide between 100 bar and 150 bar could not be seen , though. The trend that with increasing temperature the amount of water in the vapor phase increases is also found in the simulations.
T = 523.15 K, p = 200 bar - 2000 bar
In order to get an idea of the rho(p)-behaviour
of the pure (model-)components CO2 and H2O at
high pressures constant NPT simulations were carried out at T = 523 K.
As one would expect the density of pure water
is underestimated (approx. 13% at lower pressures,
around 6% at higher pressures) by the SPC model. Although the SPCE model
shows better agreement with the liquid densities
the SPC model was chosen for representing water since it gives the better
vapor pressures (see also results for H2O).
Again, the EPM2 model gives good agreement
with the experimental densities.
In the system CO2-H2O at some pressure the vapor
phase becomes more dense than the liquid phase. (The number density
of the vapor phase is still lower than the number density of the liquid
phase, the vapor (rich in CO2) reaches the higher density just
because of the higher molecular weight of CO2). In the simulation
the density inversion for the pure components takes place at a pressure
around 1450 bar while from experimental data one would expect this to happen
at a pressure around 1600 bar.
Although the results for the pure components are promising the investigation
of their binary at high pressures and temperature turned out to be much
more difficult than expected. GEMC-NPT runs were carried out and the results
compared with the experimental data by Tödheide
and Franck, Z. Phys. Chem. Neue Folge, 37, 387 (1963) resp.
Takenouchi and Kennedy, Am. J. Sc.,
262, 1055 (1964).
The results of the different authors are in quite good agreement only for
the solubility of CO2 in H2O but differ up to 12
mole % for the concentrations of H2O in the vapor phase.
First, several points on the phase envelope were evaluated using Gibbs0.9.
Relatively big system sizes (N = 400 - 600) were used to run under those
conditions to avoid program stops due to the fact that the length of one
of the simulation cells becomes smaller than the cutoff (because of the
high densities of the phases). The runs took up to 7 million steps.
One reason for the scattering of the above mentioned results might be that runs under these conditions need a very long equilibration period, especially when the number densities of the two simulation cells become very similar. Therefore a second attempt to calculate the phase envelope of CO2-H2O was made by running Gibbs1.11 with a smaller system size of N = 250. The runs took between 4 and 5.5 million steps.
Note: According to the potential parameters reported in Harris and Yung, J. Phys. Chem., 99, (1995) the cross parameters sigmaC-O were calculated using the Berthelot combining rule sigmaC-O=sqrt(sigmaC*sigmaO)*(1-kij) instead of using the usual Lorentz rule sigmaC-O=1/2*(sigmaC+sigmaO)*(1-lij) which lead to a slight correction of the Lorentz rule (lij=0.001036). This correction was removed in the meantime since its influence lies within the statistical uncertainties.
|
Go to: |
|
|---|---|
Last modified by Johannes, August 21st 1996
Page created by Johannes, August 1st 1996
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}