Paul Baranello
The most important part in modeling a process involving fluids is vapor-liquid phase behavior. This importance creates a need for accurate equilibria data for multicomponent systems, which can be applied to various chemical processes. For instance, to successfully design any type of separation apparatus, you will need to know how a multicomponent mixture will equilibrate. A more specific example of where you can use equilibria data is in the design of a process that removes aqueous wastes by supercritical water oxidation. Getting this data, unfortunately, is not that simple.
Over the years, various modeling techniques have been designed to describe multicomponent systems. The equation of state method is useful for simple fluids, but it has its drawbacks. It begins to run into problems for more complicated systems, such as aqueous systems at high pressures or ionic systems. Another limitation is that it relies on experimental data. Lastly, one can not extrapolate with confidence outside the range of experimental data. These problems create a need for a different methodology to find equilibrium data. Hopefully, this need can be filled be the technique of molecular simulations. It was not until recent developments that this method became practical to run on computers.
A widely-used method for two-phase molecular simulations is the Gibbs ensemble Monte Carlo method [1]. This technique will be used to find vapor-liquid equilibria data first for pure N2 and O2, and then for mixtures of these components. Then, the results will be compared to past experimental data. Finally, after finding the best parameters to fit the experimental data, runs will be done to see how accurately the parameters predict the behavior of these systems at high density and pressure in the single phase.
The first step in running a molecular simulation is making a model of the molecule or molecules you will be using. In these simulations, the Lennard-Jones potential is used to calculate the energy of the interactions.
Uij(r) = 4 × epsilonij [(sigmaij/r)12 - (sigmaij/r)6] (1)Where U is the configurational energy of interaction between the centers of two beads i and j a distance r apart. Epsilonij and sigmaij are energy and size parameters for the ij interaction. Sigma has units of length which represents the diameter of the bead. Epsilon has units of energy, and represents the depth of the potential well. When dealing with diatomics a third parameter, l*, is needed to describe the molecule. This is the bond length of the diatomic divided by sigma. The united atom model builds complex molecules particle by particle. For two diatomic molecules, there are four interactions between four beads instead of the simple case of one interaction between two beads. These forces must be summed to get the energy of interaction between the molecules. For interactions between different types of beads, the Lorentz-Berthelot combining rules are used.
sigmaij = (sigmaii + sigmajj) / 2 (2)
epsiolnij = (epsiolnii × epsilonjj)1/2 (3)
This procedure can be expanded to describe a system made up of hundreds of diatomics. Although their are other models that describe actual intermolecular potential better, the Lennard-Jones model is much simpler and there is a lot of literature on its properties to use for comparison to simulations.
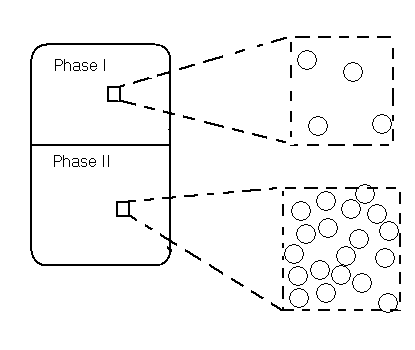
Before discussing the algorithm for doing the Gibbs ensemble calculation, let us look at the principle behind it. Figure 1 shows a macroscopic two-phase region which is in equilibrium. The procedure will be performed on a microscopic section in each of the regions. Thermodynamics gives the following requirements for equilibrium: each region is in internal equilibrium and the temperature, pressure, and chemical potential of all components in both phases are equal. First, we will deal with the pure system case and then we will move on to multi-component mixtures.
Phase I will be the gas phase and Phase II will be the liquid phase. The pure system will have constant temperature T, total volume V (= VI + VII) , and total number of particles N (= NI + NII). For this reason it will be referred to as a constant NVT system. In the pure component case, temperature is set before the simulation is performed. The other three thermodynamic requirements are satisfied by three types of moves (Figure 2): displacement of molecules within each region, volume changes of the two regions, and particle transfers between the regions. These moves satisfy internal equilibrium, equality of pressure and equality of chemical potentials respectively.
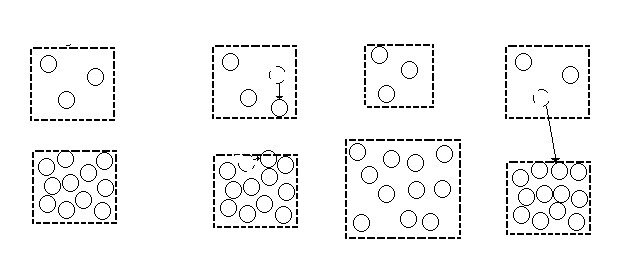
Figure 2. Types of moves.
The simulation runs as follows. After entering in all the necessary parameters (temperature, number of particles initially in each phase, initial densities, epsilon, sigma, the bond lengths), the program starts by generating an initial configuration. Next, it picks a type of move to perform. The percentage of how often a move is picked is set by the user (example: 30% displacement, 1% volume change, 69% transfer). Then, the computer attempts to make that move. The probability of the move being accepted is calculated by one of the following formulas, where Ptransfer is the formula for a transfer from region II to region I.
Pdisplacement = min [ 1 , exp(-beta × delta U) ]
Pvolume = min [ 1 , exp(-beta × delta UI - beta × delta UII + NIln ( VI + DV ) + NII ln( VII - delta V ) ]
Ptransfer = min [ 1 , NII × VI / ( (NI + 1) × VII ) × exp( -beta × delta UI -beta × delta UII ]
The computer generates a random number between zero and one. If this number is greater than the probability, then the move is rejected and the system retains the old configuration and a new move is attempted. If it is less than or equal to the number than the move is accepted and the system moves to the new configuration. This cycle is repeated on the order of 106 times. There are two periods of the simulation - the equilibration and production periods. The simulation records certain properties of the system (pressure, density, internal energy) and averages them over the production period to use for the equilibrium data at that temperature.
There are only two changes in the algorithm for multicomponent systems. First, the Ptransfer equation is done for each type of component. For example, if a system containing i and j is studied then there is one Ptransfer using NI, i and NII, i and another one with NI, j and NII, j. This procedure can be extrapolated to any number of components. The second change is the Pvolume. Once you have a second component you gain an additional degree of freedom, so for mixture simulations the pressure is also specified along with temperature before the simulation begins. Mixtures are simulated with NPT runs; total volume is no longer constant. The new equation for the probability of a volume change is:
Pvolume = min [ 1 , exp(-beta × delta UI - beta × delta UII + NI × ln ( VI + delta VI ) / VI + NII × ln( VII - delta VII ) / VII - beta × P × (VI +VII) ] (7)
There is an upper and lower temperature limit that can be used for each system When the temperature is too low, the liquid phase begins to get very dense and the chemical potential equilibration is difficult to satisfy because most of transfer moves are rejected. A particle has trouble moving from gas to liquid because most of the attempted transfers place it in a position overlapping another particle. Moving a particle from liquid to gas is difficult because it involves a large cost in energy. The upper limit is the critical point. Once the temperature of the run gets too close to the critical point, the length of interaction between molecules approaches infinity. A way of estimating the critical point is using the rectilinear diameter rule and the scaling relationship for the width of the coexistence curve.
( rholiq + rhogas )/2 = rhoc + A(Tc -T)
rholiq - rhogas = A(Tc - T)beta
A = constant; beta = 0.326The results section is divided into three parts - nitrogen, oxygen, and nitrogen-oxygen mixtures. Nitrogen will be covered in greater detail than oxygen, since the methods are repeated.
Eight runs were done for the pure nitrogen system at 5 degree intervals using the Gibbs Ensemble Program. The parameters used were [2], where kB is Boltzman’s constant:
sigma = 3.310 Angstroms epsilon / kB = 37.3 K l* = 0.329
The runs became unsuccessful under 85 K because there were not enough phase transfers. At 85 K, 0.09 % of transfer moves were being accepted. The runs were performed only up to 120 K since the critical point is 126.200 K. The liquid and vapor phases were initially set up with densities of 500 kg m-3 and 50 kg m-3, respectively. The first two million steps of the simulation were used to equilibrate the system and the next two million were for the production period. In order to check that the system had equilibrated before the production period began, the density of both the liquid and vapor phase were plotted out. Figure 3 shows an example of the liquid density.
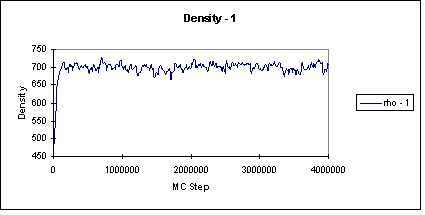
Click here for a table of the results of the nitrogen runs. After the runs were performed, they were compared to experimental data [3] in Figure 4. The graph is a little deceiving. At low temperatures, it appears that vapor simulation points overlap with the experimental points, but they do not. The Gibbs runs always under predicted the vapor density. On the liquid side, the simulation accurately predicts the density at lower temperatures until 100 K. Over 100 K, it begins to over predict the density of the liquid.
In order to predict the actual phase behavior more accurately, the parameters of the simulation were changed. This was done by putting the results and experimental data in reduced units and then adjusting the values of epsilon and sigma to minimize the error. The formulas used to reduce the system were:
Treduced = T × kB/epsilon
rhoreduced = rho × (sigma)3
Preduced = P × sigma3/epsilon
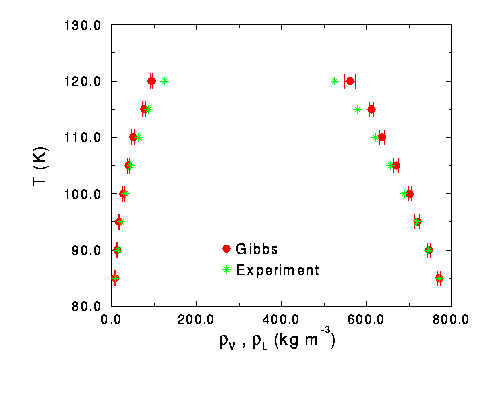
Figure 4 Phase diagram for pure nitrogen system compared with experimental results.
The simulation points were reduced with the parameters of the actual simulation. These points were then fitted to a curve using equations (2) and (3). The reduced critical properties of this fit were:
Tc* = 3.486 rhoc* = 0.2461
The experimental data was then reduced with values of s and e that would minimize the error between the reduced experimental data and the fitted curve of the simulation points. The reduced length was kept at 0.329. The best fit was with:
epsilon / kB = 36.20 K sigma = 3.299 Angstroms
These new points were then graphed in Figure 5. It must be stressed that these are not new simulation results with new parameters. They are a mathematical prediction of what the simulation results would be if the parameters were changed. One run was done for nitrogen with the new parameters at 100 K, and it accurately predicted the experimental result.
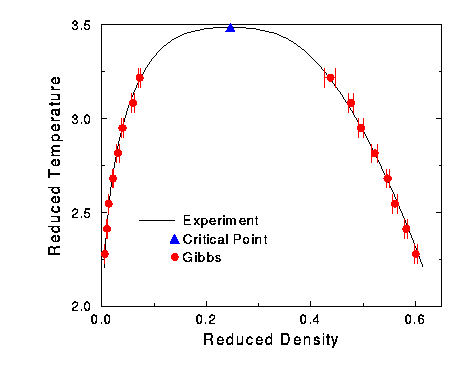
Figure 5 Phase diagram of fitted results in reduced parameters.
Another interesting property the Gibbs Ensemble Program calculates is the pressure of the boxes. Click here for a table summarizing the pressure calculations of the simulations. Once the new values of e and s were calculated, the pressure results were put in reduced form. The calculation of the vapor box is more accurate then the liquid calculation, so only the pressure calculated by the vapor is shown in Figure 6.
The test for the validity of these new Lennard-Jones parameters were to see how they predicted system behavior at high pressures and densities. NVT simulations were run with 250 particles in a box. This kept the density constant while the pressure of system was calculated. Runs were done at various densities at 120, 130, and 150 K. The simulation results were accurate at lower densities, but starting around 600 kg m-3 the simulation began predicting higher pressures than actual experimental results show. Around 800 kg m-3, the simulation had trouble creating a starting configuraton, so NVT runs above this point were not done for nitrogen.
To run simulations at denser conditions, NPT runs were performed. They were run with 250 particles in each box. Pressure was held constant while the density of the system was calculated. The simulation was set up with an originally density of 700 kg m-3. After the starting configuration was established, the density would rise above the 800 kg m-3 mark. These runs predicted experimental data quite well, and were fairly accurate even at pressures around 3000 bar.
The NPT and NVT methods were compared with one run. The NPT result was more accurate in predicting the experimental behavior at that point. Figures 7 and 8 show the isotherms of the NVT and NPT runs.
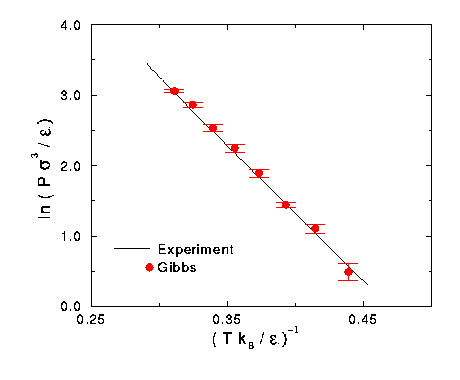
Figure 6 Vapor pressure diagram for fitted nitrogen data.
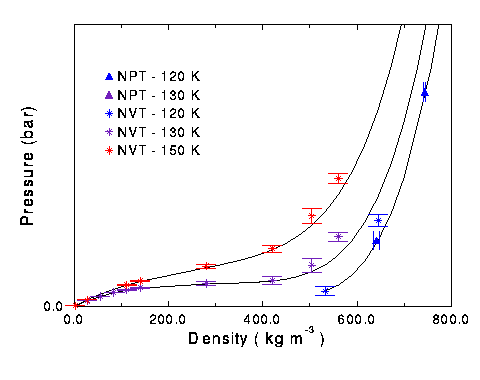
Figure 7 Isotherms of nitrogen at low pressures. Black line is experimental data.
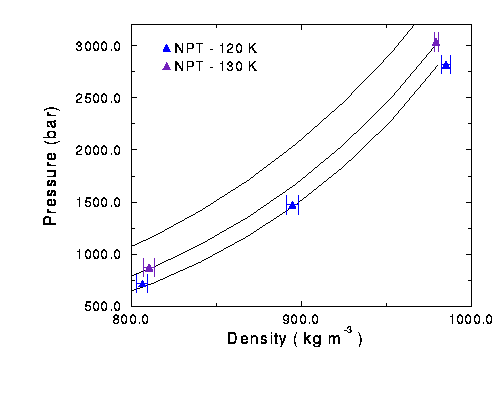
Figure 8 Isotherms of nitrogen at high pressures. Black line is experimental data.
Eleven runs were performed for pure oxygen at five degree intervals using the Gibbs ensemble program. The parameters used were[4]:
sigma = 3.090 Angstroms epsilon / kB = 44.6 K l* = 0.329
The run at 100 K was unsuccessful due to a shortage of phase transfers. It was not used in the analysis of the data. The runs were performed up to 150 K because oxygen has a critical point of 154.77 K. The liquid and vapor phases were intially set up with densities of 900 kg m-3 and 50 kg m -3, respectively. As in the nitrogen simulations, there were two million equilibration and two million production steps. Click here for a table of the two-phase region results.
The analysis performed on oxygen was the same as the one on nitrogen. The actual simulation results were plotted with experimental data (Figure 9). Once again, the simulation data correctly predicted experimental behavior at lower temperatures on the liquid side, but under predicted for the vapor density and over predicted higher temperature liquid density. So the data was put into reduced units and the s and e values were adjusted to give the best fit. The critical properties and the Lennard-Jones parameter values of this fit were:
Tc* = 3.513 rhoc* = 0.2353
epsilon / kB = 44.06 K sigma = 2.956 Angstroms
Up to this point, there was nothing significantly different between the nitrogen and oxygen runs. This changed when the vapor pressure of the fitted oxygen simulation results was graphed with the experimental results (Figure 10). All of the oxygen simulation results were above the actual experimental line. Click here for the oxygen pressure data.
NVT and NPT runs were performed for pure oxygen systems at 150 K and various densities and the results are summarized in Figure 11. Other temperatures were not tried due to lack of time. Just like in the nitrogen runs, the oxygen NVT runs were accurate at low densities, but began to over predict the pressure at higher densities. As the density was further increased, the NVT simulation started to under predict the pressure of the actual system. NPT runs were over predicting the density. The original oxygen parameters were used in a NPT simulation in order to find the source of this discrepency. This run gave an accurate prediction of the experimental result.
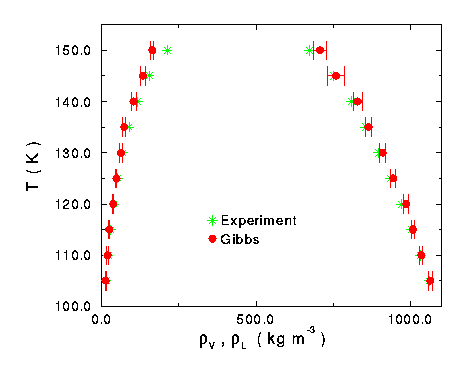
Figure 9 Phase diagram for pure oxygen system compared with experimental results.
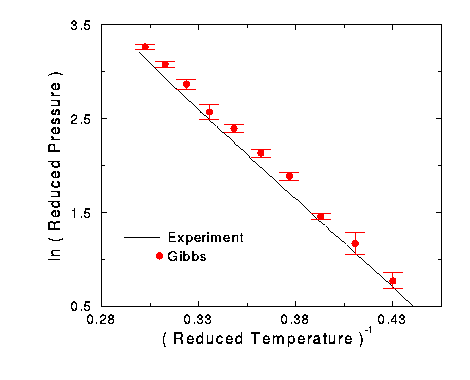
Figure 10 Vapor pressure diagram for fitted oxygen results.
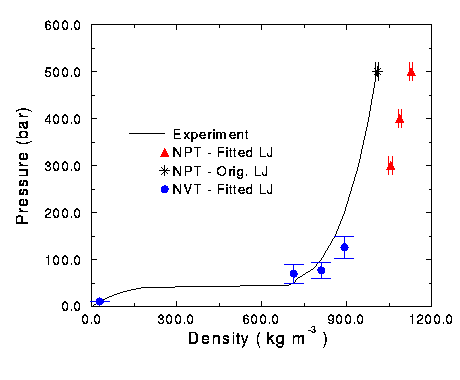
Figure 11 Isotherm of oxygen at 150 K.
Four NPT runs of different mixtures of nitrogen and oxygen were
performed. The Lennard-Jones parameters used were:
Nitrogen: sigma = 3.2987 Angstroms
epsilon / kB = 36.2 K
l* = 0.329
Oxygen: sigma = 2.9569 Angstroms
epsilon / kB = 44.1 K
l* = 0.329
Oxygen’s parameters were slightly different than the fitted parameters
described in the above section. A slight miscalculation was found in the
original oxygen fit of the experimental two-phase region data. This
miscalculation was found after the mixture runs were done, but before the
isotherm runs. Lack of time prevented the mixture runs from being redone with
the true fitted parameters, but they are still worth discussion.
All four runs were done at 100 K, with between two and a half and three
million equilibration steps and two million production steps. Not only did the
density have to equilibrate for the mixture runs, but the mole fractions in
each box had to also. The simulation results were graphed with experimental
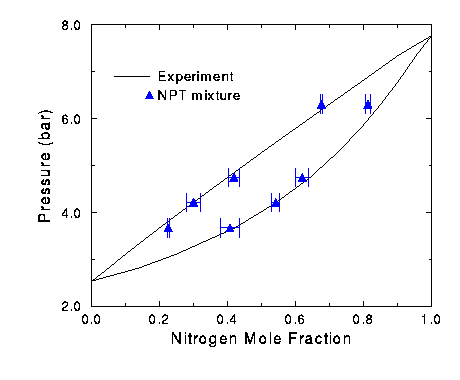
data [5] (Figure 12). The vapor side has accurate results, except for the run
at 6.3 bar. The simulation was less successful predicting the liquid line,
giving some accurate points, but also giving some results above and below the
experimental line.
Figure 12 Phase diagram for nitrogen oxygen mixture. The Gibbs Ensemble Program was written by V. Harismiadis, J. Vorholz,
A. Panagiotopoulos, and J. Errington.
[1] Panagiotopoulos, A.Z. "Direct determination of phase coexistence
properties of fluids by Monte Carlo simulation in a new ensemble,"
Molecular Physics 61, 813-826 (1987).
[2] G. Galassi and D.J. Tildesley , "Phase Diagrams of Diatomic Molecules:
Using the Gibbs Ensemble Monte Carlo Method" Molecular Simulations, 13,
11 (1994).
[3] IUPAC, volume 6, table 4, 230-232.
[4] J.E. Coon, S.Gupta, and E. McLaughlin , Chemical Physics
113, 43-52 (1987).
[5] DECHEMA Chemistry Data Series, Vapor-Liquid Equilibria for Mixtures of
Low Boiling Substances, 281.
Nitrogen-Oxygen Mixtures
Acknowledgements:
References: